

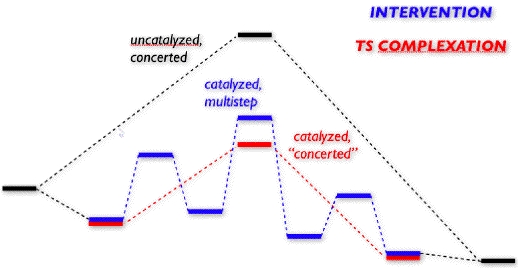
We are applying the "transition state complexation" concept to the design of transition metal-based catalysts for hydrocarbon rearrangements. The concept of accelerating a single-step (i.e. concerted) reaction by binding to its transition state structure more tightly than to the reactant(s) has a long history, yet this concept has been underexploited in the field of organometallic catalyst design. We are currently using this principle to design catalysts for metal-promoted sigmatropic shifts.
Many examples exist of transition metal-based catalysts that accelerate pericyclic reactions, but most of these examples involve a change in mechanism from a concerted rearrangement to a multistep process. We refer to the change from a concerted to a stepwise process as "intervention" by the transition metal-based catalyst, but we are focusing on avoiding this sort of mechanistic change - and the avenues to byproducts that it opens up - by exploiting the transition state complexation process, a process in which the transition state structure for the uncatalyzed reaction maintains its structure (at least approximately) and functions as a ligand for the transition metal-based catalyst (or stoichiometric "promoter").
We have previously described the effects of complexing transition state structures for electrocyclization reactions by protons (thereby turning them into sigmatropic shifts; a concept that we refer to as "transition state protonation"). We have also applied the transition state complexation idea to the promotion of otherwise orbital symmetry-forbidden 4-electron electrocyclic reactions (cyclobutene ring-openings) via complexation with iron carbonyls, and we have predicted that some [3,3] sigmatropic shifts can be accelerated through weak but selective interactions between transition state structures and halogens.
Tantillo, D. J.; Hoffmann, R. Helv. Chim. Acta 2001, 84, 1396-1404: "Demoniac Intervention in the Thermal Electrocyclic Ring- Opening of Cyclobutenes: Fe(CO)3-Complexation of Pericyclic Transition Structures," part of a special issue in honor of Edgar Heilbronner.
Hoffmann, R.; Tantillo, D. J. Angew. Chem. Int. Ed. 2003, 42, 5877-5882: "Breaking Down Barriers: The Liaison Between Sigmatropic Shifts, Electrocyclic Reactions and 3-Center Cations"
Wang, S. C..; Tantillo, D. J. J. Phys. Chem. A 2007, 111, 7149-7153: "Selective Stabilization of Transition State Structures for Cope Rearrangements of Semibullvalene and Barbaralane through Interactions with Halogens"
Siebert, M. R.; Tantillo, D. J. J. Am. Chem. Soc. 2007, 129, 8686-8687: "Transition State Complexation in Palladium-Promoted [3,3] Sigmatropic Shifts"
Matthew R. Siebert and Dean J. Tantillo: "Transition State Complexation in the Pd(II)-Catalyzed Cope Rearrangement." Lecture presented by Matt Siebert at the 237th ACS National Meeting, Salt Lake City, UT, March 22-26, 2009; paper ORGN 474.