

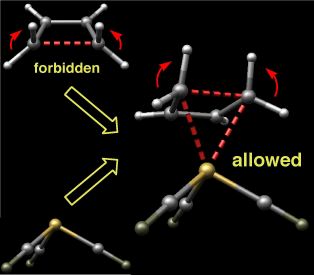
The thermal ring-opening of cyclobutene to produce butadiene is one of the prototypical examples of electrocyclic ring-opening. This reaction has been studied extensively by both experimentalists and theoreticians who have shown that it proceeds through a conrotatory transition state that is favored over alternative disrotatory or diradical pathways by at least 15 kcal/mol. These observations are in accord with predictions based on the Woodward-Hoffmann rules.
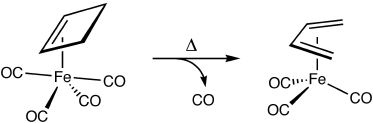
Intrigued by the possibility of altering the stereochemical course of electrocyclic processes through metal complexation, we undertook computational studies on the ring-opening of cyclobutene complexed to iron carbonyls. Density functional calculations reveal that although the disrotatory process in the absence of the organometallic fragment is symmetry forbidden, it becomes the favored pathway upon complexation due to orbital interactions that allow aromatic character to increase as the reaction proceeds.
Calculations on the related ring-opening of Fe(CO)3-methylenecyclopropane to produce Fe(CO)3-trimethylenemethane demonstrate that disrotatory ring-opening is also favored in this system. However, the favored transition state in this case involves carbon-carbon bond breaking away from the metal. Our calculations reveal the structural details of the competing disrotatory and conrotatory transition states and validate predictions made by the Carpenter group two decades ago regarding the stereoselectivity of this reaction.
Tantillo, D. J.; Hoffmann, R. Helv. Chim. Acta 2001, 84, 1396-1404: "Demoniac Intervention in the Thermal Electrocyclic Ring- Opening of Cyclobutenes: Fe(CO)3-Complexation of Pericyclic Transition Structures," part of a special issue in honor of Edgar Heilbronner.
Tantillo, D. J.; Hoffmann, R. J. Am. Chem. Soc. 2001, 123, 9855-9859: "Complicated Goings-on in the Metal-Manipulated Ring-Opening of Cyclobutene"
Tantillo, D. J.; Carpenter, B. K.; Hoffmann, R. Organometallics 2001, 20, 4562-4564: "Disrotatory and Conrotatory Transition Structures for the Fe(CO)3-Templated Rearrangement of Methylenecyclopropane to Trimethylenemethane"
Dean J. Tantillo and Roald Hoffmann: "Metal Manipulated Cyclobutene Ring-Opening Reactions." Poster presented at the 221st ACS National Meeting, San Diego, CA, April 1-5, 2001; paper ORGN 360.